

En la actualidad, se conocen los beneficios que las mitocondrias generan para el sistema celular pues le proporcionan toda la energía -inicialmente, nutrientes- necesaria de cara a su supervivencia y actividad diaria. No obstante, este microorgánulo celular eucariota contiene elementos que contribuyen al desarrollo de afecciones. Una de ellas guardan relación con la degenración muscular, las cuales se vinculan con hasta 30 clases diferentes de trastornos.
Precisamente, este lunes se han conocido los resultados de un estudio que trata el papel de un componente mitocondrial en el progreso de la distrofia muscular (DM). En este trabajo, científicos del Centro Médico del Hospital Infantil de Cincinnati (Estados Unidos) han descubierto una nueva manera de reducir los síntomas y los casos de dicho desgaste muscular, relacionada con una tipología de poros localizados en la membrana protectora de las mitocondrias que se originan cuando estas se exponen a estrés oxidativo o a una sobrecarga patológica de iones de calcio (Ca2+), la cual hace estallar el orgánulo y las fibras musculares.
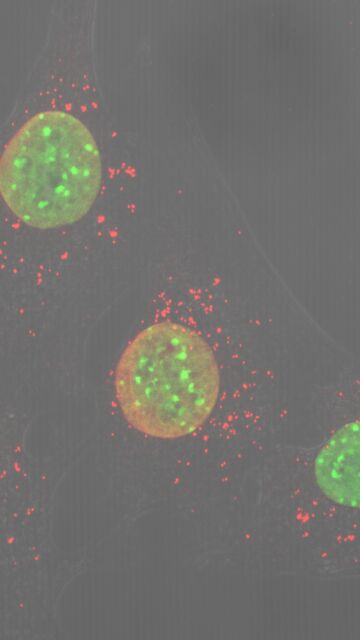
La investigación, disponible en la revista científica estadounidense 'Science Advances', ha requerido del uso de ratones editados genéticamente para demostrar que los poros de transición de permeabilidad mitocondrial (MPTP) son "el principal componente causante de la distrofia muscular", en palabras del director del estudio Jeffery Molkentin. De tal manera, han determinado que impedir su funcionamiento traería la casi completa desaparición de esta enfermedad distrófica. Al mismo tiempo, han calculado que la protección superaría el año en modelos animales (p.e. de ratón), y los 40 años en humanos.
El propio Molkentin, quien ha analizado las distrofias musculares durante más de dos décadas, ha añadido que esta aportación aún no existiría si los genes Slc25a4 y Ppif no se hubiesen extraído de los ratones, los cuales controlan la formación de los poros MPTP. Unos poros que además agilizan el padecimiento de infartos y de enfermedades neurodegenerativas tales como el Huntington, el Alzheimer o la esclerosis lateral amiotrófica (ELA).
Por ello, a lo largo de la investigación el doctor estadounidense ha destacado la importancia de controlar ambos tipos de genes para detener la llegada de las DM, y de encontrar más fármacos que motiven la cura de la distrofia muscular. Hasta ahora solo está disponible la ciclosporina A para bloquear la proteína CypD del gen Ppif; de momento ninguna acerca de la proteína tóxica ANT1 del gen Slc25a4. "También, se necesitaría más investigación para determinar si el enfoque protector de las mitocondrias protegería contra el daño cardíaco relacionado con la DM u otras disfunciones orgánicas", concluye este experto en la ciencia básica de la función y formación de las células musculares.
